του Αντώνιου Κολοκούρη, Καθηγητή του Τομέα Φαρμακευτικής Χημείας του Τμήματος Φαρμακευτικής του ΕΚΠΑ
Short summary: The papain-like protease (PLpro) of SARS-CoV-2 is a validated antiviral drug target. In a recent study Dr Jun Wang (University of Arizona) in collaboration with Prof Kolocouris (Dept of Pharmacy, NKUA) 1 (a) identified new non-covalent inhibitors of PLpro and investigated with X-ray crystallography and MD simulations the way they interacted with this enzyme (b) invented a cell-based FlipGFP reporter assay was developed for the screening of SARS-CoV-2 papain-like protease inhibitors and was shown to have a positive correlation with antiviral activity (published in ACS Cent. Sci. 2021, 7, 7, 1245–1260, IF = 15).
Extended Summary: Given the tremendous impact of the COVID-19 pandemic, the SARS-CoV outbreak in 2003 was a dire warning that was gravely overlooked in retrospect. It is imperative that therapeutics are developed that are not only effective against SARS-CoV-2 but against future strains of similar coronaviruses. The papain-like protease (PLpro) of SARS-CoV-2 is a validated and high-profile drug target, partially because it is highly conserved between SARS-CoV and SARS-CoV-2, sharing 83% sequence similarity. PLpro is involved in the cleavage of viral polyproteins and antagonizing host innate immune response through its deubiquitinating and deISG15ylating activities, rendering it a high profile antiviral drug target.
The majority of the cysteine protease inhibitors are covalent inhibitors targeting the catalytic cysteine,2 and it remains a challenge to develop noncovalent cysteine protease inhibitors with a similar potency as the covalent inhibitors.
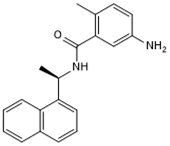
Previous attempts to discover noncovalent SARS-CoV-2 PLpro inhibitors through High-Throughput Screening (HTS) have failed to identify hits with improved enzymatic inhibition and cellular antiviral activity. 3 Through a FRET-based high-throughput screening, several hits were identified, eg. GRL0617 4 (EC50 = 25.1 µM) (Fig. 1) as PLpro inhibitors with IC50 values at the double or single-digit micromolar range. Inhibitors like GRL0617 are equally effective against both viruses, with a Ki of 0.49 μM and 0.57 μM, against SARS-CoV PLpro and SARS-CoV-2 PLpro. 4 Likewise, all critical active site residues that interact with GRL0617 are conserved. These similarities would indicate that PLpro inhibitors might retain their activity against beta coronaviruses that might emerge in the future. While among the reported SARS-CoV or SARS-CoV-2 PLpro inhibitors, GRL0617 is one of the most potent compounds, it had weak antiviral activity (Vero E6: EC50 = 23.64 μM; Caco2-hACE2: EC50 = 19.96 μM). Structural analogues of GRL0617 were also designed and synthesized; however, none showed improved enzymatic inhibition. 5
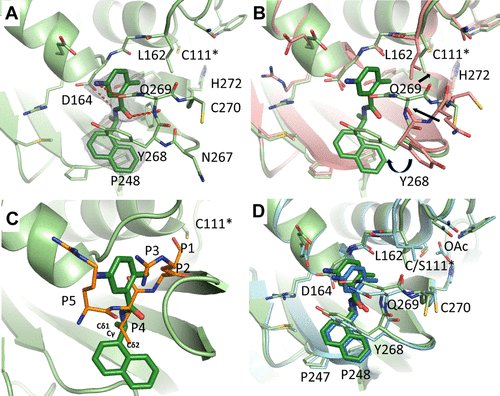
In this study we solved the X-ray crystal structure of the wild-type SARS-CoV-2 PLpro in complex with GRL0617 (Fig. 1). Binding of GRL0617 to SARS-CoV-2 induced a conformational change in the BL2 loop to the more closed conformation. Part of the reason for the difficulty in targeting SARS-CoV-2 PLpro is the lack of S1 and S2 pockets (Fig. 1), which leaves only S3 and S4 pockets for inhibitor binding. The intrinsic flexibility of the BL2 loop implies that structurally diverse inhibitors might be able to fit in the S3–S4 pockets.
We aimed to identify more potent SARS-CoV-2 PLpro inhibitors through a HTS. To prioritize lead compounds for the cellular antiviral assay against SARS-CoV-2, we developed a cell-based fluorescence resonance energy transfer (FlipGFP assay) that is suitable for quantifying the intracellular enzymatic inhibition potency of PLpro inhibitors. The advantage of the FlipGFP-PLpro assay over the standard FRET-based enzymatic assay is that it can rule out compounds that are either cytotoxic or membrane impermeable or nonspecifically modifying the catalytic cysteine through oxidation or alkylation.
Through this FlipGFP-PLpro assay high-throughput screening two compounds selected from the FlipGFP-PLpro assay, Jun9-53-2 and Jun9-72-2, inhibited SARS-CoV-2 replication in Caco-2 hACE2 cells with EC50 values of 8.89 and 8.32 µM, respectively, which were 3-fold more potent than GRL0617 (EC50 = 25.1 µM). The X-ray crystal structures of PLpro in complex with GRL0617 showed that binding of GRL0617 to SARS-CoV-2 induced a conformational change in the BL2 loop to the more closed conformation (Fig. 1). After subsequent lead optimization, we identified several PLpro inhibitors including Jun9-72-2 and Jun9-75-4 with improved enzymatic inhibition and antiviral activity compared to GRL0617, which was reported as a SARS-CoV PLpro inhibitor.
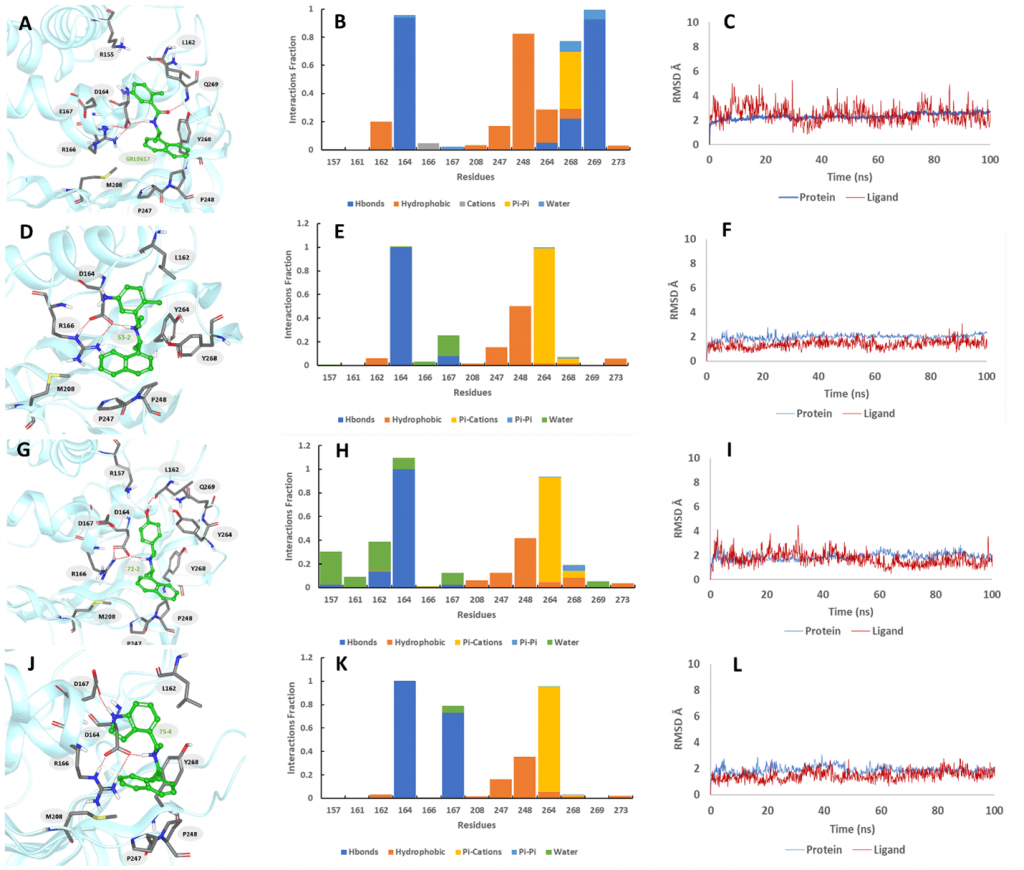
In total, three PLpro inhibitors Jun9-72-2, Jun9-85-1, and Jun9-87-1 were identified as potent SARS-CoV-2 antivirals with EC50 values at or less than 10 μM when tested in both the Vero E6 and Caco2-hACE2 cell lines (Fig. 2). As shown by the MD simulations (Fig. 3), the replacement of the carboxamide group in GRL0617 to the trialkyl ammonium in Jun9-53-2, Jun9-72-2, and Jun9-75-4 affects the binding interactions inside the receptor-binding region. In comparison to GRL0617, the N–H+ group in Jun9-53-2, Jun9-72-2, and Jun9-75-4 is engaged in strong ionic hydrogen bonding interactions with a side chain of Asp164, participating in another stabilizing ionic hydrogen bonding interactions with Arg166, which pulls the ligands inside the receptor-binding region from the hydrogen-bonded Gln269 to a new T-shaped π–π stacking with the Ty264 instead of Tyr268 in GRL0617. All the four ligands form hydrophobic interactions between naphthalene ring and Pro248. In conclusion, the SARS-CoV-2 PLpro inhibitors discovered in this study represent promising hits for further development as SARS-CoV-2 antivirals, the FlipGFP-PLpro assay is a suitable surrogate for testing the cellular activity of PLpro inhibitors in the BSL-2 setting, and the results can be used to help prioritize leads for the antiviral assay. This is expected to speed up the drug discovery process.

References
(1) Ma, C.; Sacco, M. D.; Xia, Z.; Lambrinidis, G.; Townsend, J. A.; Hu, Y.; Meng, X.; Szeto, T.; Ba, M.; Zhang, X.; et al. Discovery of SARS-CoV-2 Papain-like Protease Inhibitors through a Combination of High-Throughput Screening and a FlipGFP-Based Reporter Assay. ACS Cent. Sci. 2021, 7 (7), 1245–1260. https://doi.org/10.1021/acscentsci.1c00519.
(2) Siklos, M.; BenAissa, M.; Thatcher, G. R. J. Cysteine Proteases as Therapeutic Targets: Does Selectivity Matter? A Systematic Review of Calpain and Cathepsin Inhibitors. Acta Pharmaceutica Sinica B. 2015. https://doi.org/10.1016/j.apsb.2015.08.001.
(3) Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Zhu, W.; Cao, D.; et al. The Complex Structure of GRL0617 and SARS-CoV-2 PLpro Reveals a Hot Spot for Antiviral Drug Discovery. Nat. Commun. 2021. https://doi.org/10.1038/s41467-020-20718-8.
(4) Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M.; Baliji, S.; Chaudhuri, R.; Fu, W.; Prabhakar, B. S.; Johnson, M. E.; et al. A Noncovalent Class of Papain-like Protease/Deubiquitinase Inhibitors Blocks SARS Virus Replication. Proc. Natl. Acad. Sci. U. S. A. 2008. https://doi.org/10.1073/pnas.0805240105.
(5) Osipiuk, J.; Azizi, S.-A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K. A.; Kang, S.; Kathayat, R. S.; Kim, Y.; Lisnyak, V. G.; et al. Structure of Papain-like Protease from SARS-CoV-2 and Its Complexes with Non-Covalent Inhibitors. Nat. Commun. 2021, 12 (1), 743. https://doi.org/10.1038/s41467-021-21060-3.