


Μαρία Τζέτη: Καθηγήτρια Γενετικής, Ιατρική Σχολή ΕΚΠΑ
Μυρτώ Πούλου: Ακαδημαϊκός Υπότροφος
Θωμάς Μπραντζος: Επιστημονικός συνεργάτης
Ι. Ρ. Συνοδινού-Traeger: Καθηγήτρια Γενετικής, Διευθύντρια
Εργαστήριο Ιατρικής Γενετικής, Ιατρική Σχολή ΕΚΠΑ
H νόσος Wilson ή ηπατοφακοειδής εκφύλιση είναι μια σπάνια κληρονομική νόσος που περιγράφηκε για πρώτη φορά το 1912 από τον Άγγλο νευρολόγο Samuel Alexander Kinnier Wilson. Μεταβιβάζεται με υπολειπόμενο αυτοσωμικό χαρακτήρα και οφείλεται στην αδυναμία αποβολής του χαλκού και την προοδευτική συσσώρευσή του σε ιστούς, όπως στο ήπαρ, και στη συνέχεια στον εγκέφαλο, νεφρό και κερατοειδή χιτώνα (Εικ.1) [1].
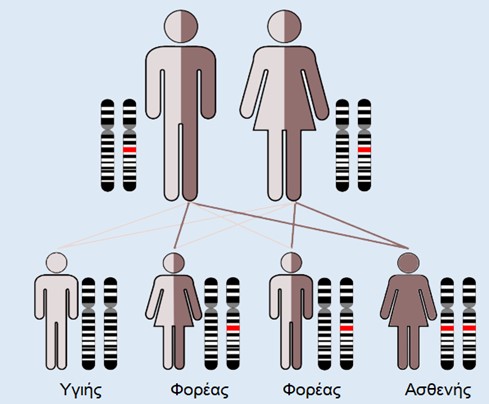
Παγκοσμίως η συχνότητα νόσου εκτιμάται 1 στις 30000 γεννήσεις και η συχνότητα φορέων 1 στους 90. Στη χώρα μας αναμένεται κάθε χρόνο η γέννηση 1 έως 5 νέων ασθενών, ενώ σε κλειστές κοινωνίες, όπως αυτές της ορεινής Κρήτης και της Καλύμνου, η συχνότητα παρουσιάζεται σημαντικά αυξημένη. H ηλικία έναρξης των συμπτωμάτων κυμαίνεται από την 1η μέχρι την 10η δεκαετία ζωής.
Συμπτώματα-Επιπλοκές
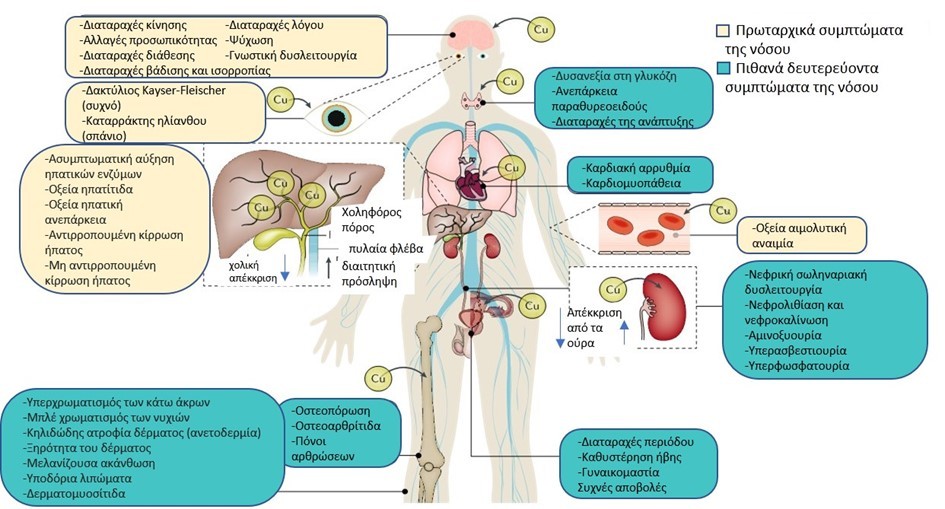
Οι κλινικές εκδηλώσεις οφείλονται στην τοξικότητα του χαλκού και ποικίλλουν ανάλογα με το όργανο που προσβάλεται. Η εξέλιξη της νόσου συνδέεται με βλάβη του ήπατος και συμπτωματολογία αντίστοιχη της ηπατικής και νεφρικής ανεπάρκειας, καθώς και νευροψυχιατρική συμπτωματολογία (Εικ.2) [4].
Παθοφυσιολογία
Η νόσος προκαλείται από παθογόνες παραλλαγές του υπεύθυνου γονιδίου ATP7B το οποίο εντοπίζεται στο χρωμόσωμα 13q14.3 [g.51932669-52012130; GRCh38.p13) και κωδικοποιεί μια αδενοσινοτριφωσφατάση τύπου Ρ -ATPase2. Εντοπίζεται στο σωματίδιο Golgi των ηπατοκυττάρων και είναι υπεύθυνη για την ενσωμάτωσή του χαλκού στη σερουλοπλασμίνη και την απόδοση της περίσσειας χαλκού στη χολή για απέκκριση. Εάν τα επίπεδα χαλκού στο ήπαρ είναι πολύ υψηλά, η ΑΤΡase2 μεταφέρεται από το σωματίδιο Golgi σε κυστίδια για την αποβολή περίσσειας χαλκού μέσω της χολής.
Στη νόσο Wilson οι παθογόνες παραλλαγές οδηγούν σε μειωμένη λειτουργικότητα της ATP7B, ενώ η σερουλοπλασμίνη παράγεται σε μια ανενεργή μορφή χωρίς δέσμευση χαλκού (αποσερουλοπλασμίνη) και αποσυντίθεται ταχέως στο αίμα. Ο χαλκός συσσωρεύεται σταδιακά στον ηπατικό ιστό και μόλις ξεπεράσει τις αποθηκευτικές του ικανότητες απελευθερώνεται στο αίμα και εναποτίθεται σε επιπλέον ιστούς.
Διάγνωση
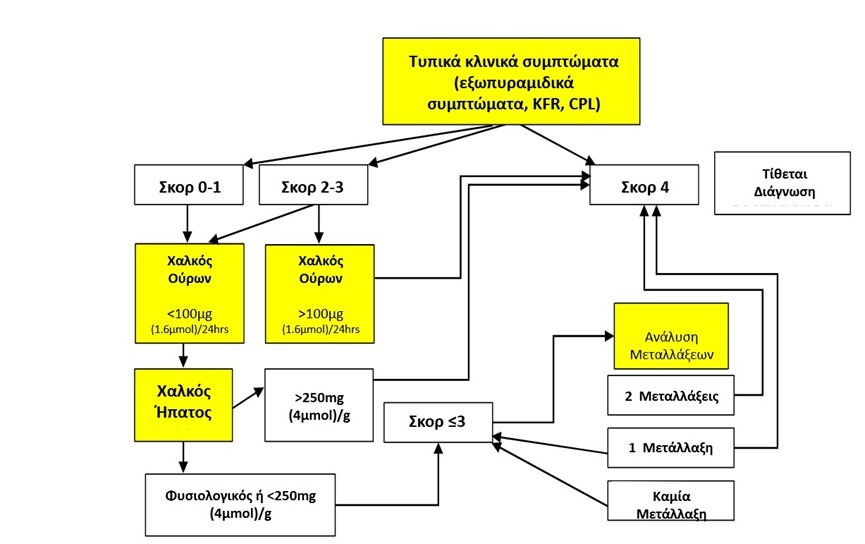
Η νόσος Wilson εμφανίζει παρόμοιο φαινότυπο με άλλες ηπατικές ή νευρολογικές διαταραχές, καθιστώντας δύσκολη τη διάγνωση. Συνιστάται ένας συνδυασμός βιοχημικών και οφθαλμολογικών εξετάσεων, βιοψίας ήπατος και γενετικού ελέγχου. Ο εντοπισμός φορέων και η διάγνωση ασυμπτωματικών ασθενών με οικογενειακό ιστορικό είναι εφικτή μόνο με μοριακή ανάλυση του ATP7B.
Η διάγνωση της νόσου τίθεται, όταν ισχύουν τα κάτωθι κριτήρια:
- Χαμηλές τιμές σερουλοπλασμίνης ορού <20mg/dl
- Αύξηση αποβαλλόμενου χαλκού ούρων 24ώρου >40μg/l
- Παρουσία δακτυλίου Kayser-Fleischer στον κερατοειδή χιτώνα
- Αύξηση του περιεχομένου χαλκού στον ηπατικό ιστό >250μg/gr ξηρού ηπατικού ιστού.
Μοριακή Διαταραχή/ Μεταλλάξεις στον Ελληνικό Πλυθυσμό
Περισσότερες από 600 μεταλλάξεις υπεύθυνες για τη νόσο έχουν ανιχνευθεί μέχρι σήμερα με σαφή εθνική κατανομή (συγκεκριμένο είδος μεταλλάξεων σε διαφορετικές πληθυσμιακές ομάδες). Η πιο συχνή μετάλλαξη παγκοσμίως είναι η p.His1069Gln (H1069Q) με συχνότητα έως και 65% στην κεντρική Ευρώπη και 10-35% σε Μεσογειακές χώρες. Εκτός της κοινής σε όλους τους πληθυσμούς μετάλλαξης, το υπόλοιπο φάσμα τείνει να είναι διαφορετικό.
To Εργαστήριο Ιατρικής Γενετικής του Εθνικού και Καποδιστριακού Πανεπιστημίου Αθηνών ξεκίνησε τον μοριακό έλεγχο σε ασθενείς με υποψία νόσου Wilson το 1997 και αποτελεί το κέντρο αναφοράς για το νόσημα στην Ελλάδα έχοντας ελέγξει το σύνολο των ασθενών. Στην εικόνα 4 παρουσιάζονται οι συχνότερες μεταλλάξεις που έχουν βρεθεί σε ασθενείς Ελληνικής καταγωγής με τις συχνότητές τους. [2, 3]
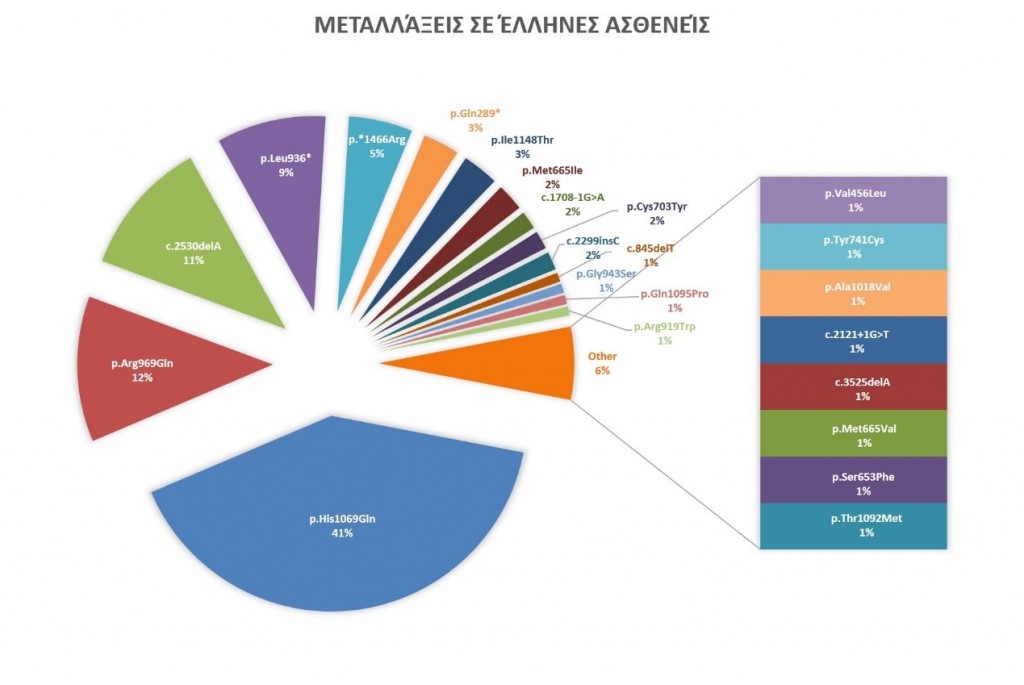
Στην Ελλάδα υπάρχει μεγάλη συγκέντρωση ασθενών με συγκεκριμένους γονοτύπους, όπως στην περιοχή των Κυκλάδων (Μήλο, Φολέγανδρο, Σίφνο) αλλά και στην Κάλυμνο. Ο πιο συχνός συνδυασμός μεταλλάξεων σε αυτές τις περιοχές είναι συνδυασμός των p.Arg969Gln και p.Ile1148Thr με την πλέον κοινή́ μετάλλαξη p.His1069Gln. Το γεγονός αυτό́ είναι αναμενόμενο και λόγω της κλειστής γονιδιακής δεξαμενής σε νησιά́ όπου αναπόφευκτα και λόγω του μικρού́ πληθυσμού́ έχουμε φαινόμενα εγκαθιδρύσεων με γάμους μεταξύ́ συγγενικών ατόμων ακόμη και εάν οι ίδιοι αγνοούν κάποια μακρινή συγγένεια.
Θεραπεία
Η εξέλιξη της νόσου Wilson χωρίς έγκαιρη διάγνωση και θεραπεία, μπορεί να προκαλέσει μακροχρόνιες βλάβες και να καταστεί θανατηφόρα. Η θεραπεία στοχεύει στην απομάκρυνση των τοξικών ποσοτήτων του χαλκού και στην αποτροπή της επανασυσσώρευσης στους ιστούς, με την χορήγηση χηλικών παραγόντων (κυρίως πενικιλαμμίνης και τριεντίνης) που δεσμεύουν τον χαλκό και οδηγούν στην απέκκρισή του από τα ούρα. Σε ασθενείς με προχωρημένη ηπατική νόσο ή οξεία ηπατική ανεπάρκεια η μόνη επιλογή θεραπείας είναι η μεταμόσχευση ήπατος.
[1] Ferenci P. Diagnosis of Wilson disease. In: Handbook of Clinical Neurology. 2017.
[2] Panagiotakaki E, Tzetis M, et al. Genotype-phenotype correlations for a wide spectrum of mutations in the Wilson disease gene (ATP7B). American Journal of Medical Genetics. 2004. [3] Panagiotakaki E, Tzetis M, et al. Genotype-phenotype correlations for a wide spectrum of mutations in the Wilson disease gene (ATP7B). Am J Med Genet. 2004;131 A(2). [4] Członkowska A, et al. Wilson disease. Nature Reviews Disease Primers. 2018.













