
Από το Εργαστήριο Ιατρικής Γενετικής, Ιατρική Σχολή, Νοσοκομείο Παίδων «Η Αγία Σοφία», ΕΚΠΑ

Νίκος Μαρινάκης MSc, PhD – Μοριακός Βιολόγος & Γενετιστής, Επιστημονικός συνεργάτης – Biocurator of the ClinGen Retina Gene Curation Expert Panel
Μαρία Τσίπη PhD – Βιολόγος

Μαρία Τζέτη, Καθηγήτρια Γενετικής

Joanne Traeger-Συνοδινού, Καθηγήτρια Γενετικής, Διευθύντρια Εργαστηρίου Ιατρικής Γενετικής
Οι κληρονομικές παθήσεις του αμφιβληστροειδούς αποτελούν μια ομάδα νοσημάτων με μεγάλη κλινική και γενετική ετερογένεια επηρεάζοντας περίπου 1 στα 2.000 άτομα παγκοσμίως. Οι συνέπειες για τα άτομα αυτά ποικίλλουν από καθολική τύφλωση στις περιπτώσεις σοβαρού εκφυλισμού του αμφιβληστροειδούς, όπως στην συγγενή αμαύρωση Leber (Leber’s Congenital Amaurosis, LCA), μέχρι ήπιες δυσλειτουργίες του αμφιβληστροειδούς (νυχτερινή τύφλωση, αχρωματοψία κ.α.). Η έκταση της βλάβης στα κύτταρα του οφθαλμού, η προοδευτική εξέλιξη και η ηλικία εμφάνισης εξαρτώνται σε μεγάλο βαθμό από τη γενετική αιτία [1, 2].
Οι ασθένειες αυτές μπορούν να ταξινομηθούν σε τέσσερις μεγάλες ομάδες: στις ασθένειες όπου προσβάλλονται κυρίως τα ραβδία, σε αυτές όπου προσβάλλονται τα κωνία, στις γενικές αμφιβληστροειδοπάθειες όπου προσβάλλονται και οι δύο τύποι φωτοϋποδοχέων και τέλος στις υαλοαμφιβληστροειδοπάθειες. Οι διάφοροι τύποι κληρονομικών αμφιβληστροειδοπαθειών μπορεί να είναι συνδρομικού ή μη συνδρομικού τύπου, με τους τελευταίους να περιλαμβάνουν κυρίως τη μελαχρωστική αμφιβληστροειδοπάθεια (Retinitis Pigmentosa, RP), τη δυστροφία κωνίων-ραβδίων, δυστροφία κερατοειδούς, συγγενή αμαύρωση Leber, συγγενή καταρράκτη, νόσο Stargardt κ.α. [1].
Τα τελευταία 20 χρόνια οι πληροφορίες σχετικά με την μοριακή βάση των αμφιβληστροειδοπαθειών αυξάνονται συνεχώς. Μέχρι σήμερα, έχουν συσχετιστεί με αυτά τα νοσήματα περισσότερα από 280 γονίδια (RetNet: https://sph.uth.edu/retnet/, Οκτώβριος 2022). Τα σχετιζόμενα αυτά γονίδια που προκαλούν αμφιβληστροειδοπάθεια μπορεί να κληρονομούνται με αυτοσωματικό υπολειπόμενο ή επικρατητικό τρόπο, ενώ κάποιες από αυτές μπορεί να κληρονομούνται και με τους δύο τρόπους. Ειδικότερα, η πλειοψηφία των ασθενών (50-60%) φέρουν αυτοσωματικές υπολειπόμενες παραλλαγές, ενώ είναι συχνές και οι οικογένειες με αυτοσωματικές επικρατητικές παραλλαγές (30-40%). Σημαντικό ποσοστό είναι επίσης και οι περιπτώσεις που παρουσιάζουν φυλοσύνδετη κληρονομικότητα (5-20%). Τέλος, μπορεί να υπάρχει και μιτοχονδριακή κληρονομικότητα (1-3%) σε ορισμένους τύπους αμφιβληστροειδοπαθειών [1, 2].
Οι μισοί περίπου ασθενείς με κληρονομικές αμφιβληστροειδοπάθειες πάσχουν από μελαγχρωστική αμφιβληστροειδοπάθεια. Η μελαγχρωστική αμφιβληστροειδοπάθεια (Retinitis Pigmentosa, RP) ανήκει στις προοδευτικές αμφιβληστροειδοπάθειες που προσβάλλουν πρώτα τα ραβδία. Η RP περιλαμβάνει μια ομάδα κλινικά όμοιων φαινοτύπων που έχουν όμως διαφορετικά γενετικά αίτια. Περισσότερες από 5000 παραλλαγές, σε περισσότερα από 60 γονίδια, σχετίζονται με τη μη συνδρομική μελαγχρωστική αμφιβληστροειδοπάθεια. Η RP είναι συνήθως μη συνδρομική, ενώ υπάρχουν και συνδρομικές μορφές, με την πιο συχνή από αυτές το σύνδρομο Usher [3].
Μία από τις πιο σοβαρές μορφές κληρονομικής αμφιβληστροειδοπάθειας είναι η Συγγενής Αμαύρωση Leber (Leber’s Congenital Amaurosis, LCA). Αντιπροσωπεύει μια ομάδα κληρονομικών παθήσεων του αμφιβληστροειδούς που χαρακτηρίζονται από σοβαρή και από μικρή ηλικία απώλεια όρασης, νυσταγμό, αμαυρωτικές κόρες και απόντα ηλεκτρικά σήματα στο ηλεκτροαμφιβληστροειδογράφημα. Στις περισσότερες περιπτώσεις αυτή η νόσος κληρονομείται με αυτοσωματικό υπολειπόμενο τρόπο. Μέχρι στιγμής, περισσότερα από 16 γονίδια έχει βρεθεί ότι εμπλέκονται στην εμφάνιση της νόσου. Σήμερα, η LCA θεωρείται ως η πιο σοβαρή δυστροφία του αμφιβληστροειδούς χωρίς να αποτελεί συστηματική ασθένεια [4].
Οι πολλοί και αλληλεπικαλυπτόμενοι τύποι, οι διαφορετικοί τρόποι κληρονόμησης και οι πολλαπλά σχετιζόμενοι γενετικοί τόποι καθιστούν την τελική διάγνωση των αμφιβληστροειδοπαθειών μια διαδικασία αρκετά επίπονη, χρονοβόρα και με υψηλό κόστος. Ωστόσο, τα σημαντικά επιτεύγματα που έχουν σημειωθεί και η βελτίωση των μοριακών τεχνικών τα τελευταία έτη έχουν δώσει τη δυνατότητα για τον ταχύ και αξιόπιστο εντοπισμό των γενετικών αιτιών που ευθύνονται για τις κληρονομικές αμφιβληστροειδοπάθειες με στόχο την ακριβή και οριστική διάγνωση, προσφέροντας σημαντικές πληροφορίες για την πρόγνωση και εξέλιξη της νόσου, το κατάλληλο πλαίσιο για γενετική συμβουλευτική των ασθενών και των οικογενειών τους και τέλος τη δυνατότητα θεραπευτικής αντιμετώπισης.
Η εφαρμογή των τεχνολογιών αλληλούχησης επόμενης γενιάς (Next Generation Sequencing-NGS) στην κλινική πράξη για τη γενετική ταυτοποίηση των κληρονομικών αμφιβήστροειδοπαθειών έχει οδηγήσει πολλές ερευνητικές ομάδες και εταιρείες να αναπτύξουν θεραπευτικά πρωτόκολλα βασιζόμενα στη γονιδιακή θεραπεία [5]. Εδώ και αρκετά χρόνια γίνεται προσπάθεια θεραπείας των ασθενειών με γονιδιακή ενίσχυση ή αποκατάσταση. Συνήθως, γενετικές ασθένειες που οφείλονται σε μείωση ή απώλεια μιας πρωτεΐνης με συγκεκριμένη λειτουργία, αποτελούν ιδανικούς στόχους για γονιδιακή θεραπεία. Η έκφραση του φυσιολογικού γονιδίου σε φυσιολογικά επίπεδα στα κύτταρα-στόχους θα μπορούσε να οδηγήσει στην οριστική αντιμετώπιση της νόσου [6].
Στις 15 Σεπτεμβρίου 2022 πραγματοποιήθηκε η πρώτη γονιδιακή θεραπεία στην Ελλάδα σε 44χρονη ασθενή με σοβαρή απώλεια όρασης από την Α’ Πανεπιστημιακή Οφθαλμολογική Κλινική του ΓΝΑ Αθηνών «Γ. Γεννηματάς» (https://hub.uoa.gr/the-first-gene-therapy-in-greece-in-a-patient-with-severe-vision-loss/). Το φάρμακο που χρησιμοποιήθηκε γι’ αυτή τη θεραπεία περιέχει τη δραστική ουσία voretigene neparvovec και είναι ένα είδος φαρμάκου προηγμένης γονιδιακής θεραπείας. Η συγκεκριμένη θεραπεία μπορεί να χρησιμοποιηθεί μόνο όταν οι ασθενείς έχουν επαρκή αριθμό λειτουργικών αμφιβληστροειδικών κυττάρων και όταν η νόσος προκαλείται από παραλλαγές στο γονίδιο RPE65, το οποίο κληρονομείται με αυτοσωμικό υπολειπόμενο τρόπο και είναι απαραίτητο για τη φυσιολογική λειτουργία των αμφιβληστροειδικών κυττάρων [6, 7].
Γονιδιακές θεραπείες για άλλους τύπους κληρονομικών αμφιβληστροειδοπαθειών (γονίδια ABCA4, MERTK, PDE6B, RPGR, CNGB3, CEP290, USH2A, ΜΤ-ND4 κ.α.) βρίσκονται σε στάδια κλινικών δοκιμών αυτή τη στιγμή με αρκετά ενθαρρυντικά αποτελέσματα, δίνοντας τη δυνατότητα σε αυτή τη μεγάλη ομάδα ασθενών μελλοντικά να θεραπευτούν από την αντίστοιχη νόσο [8].
Απαραίτητο βήμα για την εφαρμογή της γονιδιακής θεραπείας είναι πρωτίστως η γενετική ταυτοποίηση της νόσου, δηλαδή ο εντοπισμός της αιτιοπαθογόνου παραλλαγής στο υπεύθυνο γονίδιο. Η χρήση της αλληλούχησης όλων των εξονίων του γονιδιώματος (Whole Exome Sequencing-WES) ως διαγνωστικό εργαλείο έχει αλλάξει τη στρατηγική αναλύσεων, από την αλληλούχηση μόνο των σχετιζόμενων με ένα φαινότυπο γονιδίων σε σάρωση όλων των γονιδίων που δυνητικά μπορούν να εξηγήσουν μια συγκεκριμένη διαταραχή. Η συνεχής ταυτοποίηση νέων σχετιζόμενων γονιδίων με κληρονομικές αμφιβληστροειδοπάθειες καθιστά το WES πολύτιμο και οικονομικό εργαλείο στην παρούσα φάση σε σχέση με ανάλυση μεμονωμένων γονιδίων ή πάνελ γονιδίων, δίδοντας τη δυνατότητα κάλυψης της διαφοροδιάγνωσης, αλλά και επανανάλυσης των δεδομένων μελλοντικά, ώστε να εντοπιστεί πιθανά η γενετική βλάβη σε νέα σχετιζόμενα γονίδια [5].
Το Εργαστήριο Ιατρικής Γενετικής της Ιατρικής Σχολής του Εθνικού και Καποδιστριακού Πανεπιστημίου Αθηνών (https://iatrikigenetiki.med.uoa.gr), με έδρα το Νοσοκομείο Παίδων «Η Αγία Σοφία», αποτελεί το μεγαλύτερο δημόσιο ακαδημαϊκό κέντρο, καθώς και κέντρο αναφοράς για τη γενετική αξιολόγηση και διάγνωση των σπάνιων παθήσεων στην Ελλάδα, κυρίως σε παιδιά. Τις τελευταίες 2 δεκαετίες έχει τεθεί διάγνωση στο εργαστήριο σε χιλιάδες ασθενείς με τέτοια νοσήματα.
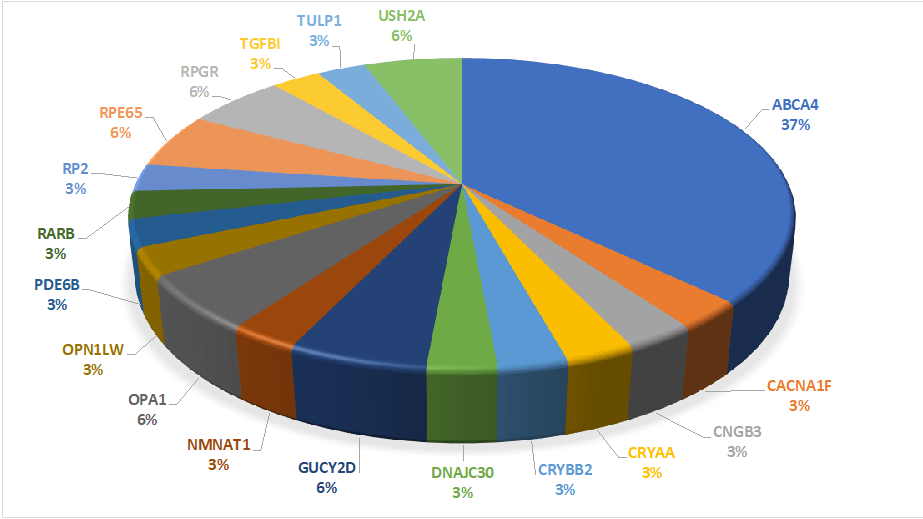
Έως σήμερα στο Εργαστήριο Ιατρικής Γενετικής έχουν διαγνωστεί 35 ασθενείς που πάσχουν από κάποιο τύπο κληρονομικής αμφιβληστροειδοπάθειας [5, 9]. Η μοριακή γενετική διάγνωση ταυτοποίησε αιτιοπαθολογικές παραλλαγές σε 18 διαφορετικά γονίδια (Εικόνα 1).
Οι παραπάνω διαγνώσεις πραγματοποιήθηκαν σε συνεργασία με εξειδικευμένους και έμπειρους οφθαλμιάτρους και κλινικούς γενετιστές. Η διεπιστημονική προσέγγιση με τη συνεργασία κλινικών και εργαστηριακών επιστημόνων αποτελεί ακρογωνιαίο λίθο για την οριστική διάγνωση των σπάνιων νοσημάτων στο Εργαστήριο Ιατρικής Γενετικής.
Βιβλιογραφία:
- García Bohórquez B, Aller E, Rodríguez Muñoz A, Jaijo T, García García G and Millán JM (2021) Updating the Genetic Landscape of Inherited Retinal Dystrophies. Front. Cell Dev. Biol. 9:645600. doi: 10.3389/fcell.2021.645600
- Ellingford, J. M., Hufnagel, R. B., & Arno, G. (2020). Phenotype and Genotype Correlations in Inherited Retinal Diseases: Population-Guided Variant Interpretation, Variable Expressivity and Incomplete Penetrance. Genes, 11(11), 1274. doi: 10.3390/genes11111274
- Koyanagi, Y., Akiyama, M., Nishiguchi, K. M., Momozawa, Y., Kamatani, Y., Takata, S., Inai, C., Iwasaki, Y., Kumano, M., Murakami, Y., Omodaka, K., Abe, T., Komori, S., Gao, D., Hirakata, T., Kurata, K., Hosono, K., Ueno, S., Hotta, Y., Murakami, A., … Sonoda, K. H. (2019). Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. Journal of medical genetics, 56(10), 662–670. https://doi.org/10.1136/jmedgenet-2018-105691
- Kumaran, N., Moore, A. T., Weleber, R. G., & Michaelides, M. (2017). Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. The British journal of ophthalmology, 101(9), 1147–1154. https://doi.org/10.1136/bjophthalmol-2016-309975
- Marinakis, N. M., Svingou, M., Veltra, D., Kekou, K., Sofocleous, C., Tilemis, F. N., Kosma, K., Tsoutsou, E., Fryssira, H., & Traeger-Synodinos, J. (2021). Phenotype-driven variant filtration strategy in exome sequencing toward a high diagnostic yield and identification of 85 novel variants in 400 patients with rare Mendelian disorders. American journal of medical genetics. Part A, 185(8), 2561–2571. doi: 10.1002/ajmg.a.62338
- Nuzbrokh, Y., Ragi, S. D., & Tsang, S. H. (2021). Gene therapy for inherited retinal diseases. Annals of translational medicine, 9(15), 1278. doi: 10.21037/atm-20-4726
- Sodi, A., Banfi, S., Testa, F., Della Corte, M., Passerini, I., Pelo, E., Rossi, S., Simonelli, F., & Italian IRD Working Group (2021). RPE65-associated inherited retinal diseases: consensus recommendations for eligibility to gene therapy. Orphanet journal of rare diseases, 16(1), 257. doi: 10.1186/s13023-021-01868-4
- Georgiou, M., Fujinami, K., & Michaelides, M. (2021). Inherited retinal diseases: Therapeutics, clinical trials and end points-A review. Clinical & experimental ophthalmology, 49(3), 270–288. doi: 10.1111/ceo.13917
- Tsipi, M., Tzetis, M., Kosma, K., Moschos, M., Braoudaki, M., Poulou, M., Kanavakis, E., Kitsiou-Tzeli, S. (2016). Genomic Screening of ABCA4 and arrayCGH analysis underline the genetic variability of Greek patients with inherited retinal diseases. Meta Gene, 8, 37-43.